Conformational entropy estimation by Gaussian mixtures
|
Gergely Gyimesi, Péter Závodszky, and András Szilágyi: |
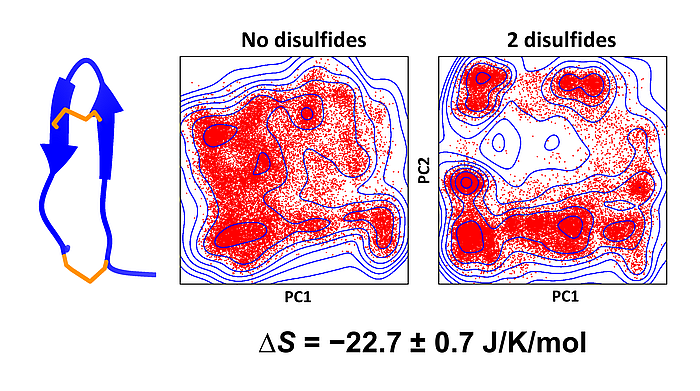
We have developed a novel method to estimate configurational entropy of biomolecules from conformational ensembles generated by simulations or experiment. The method is based on estimating the probability density function by fitting a Gaussian mixture onto the ensemble of coordinates. We have shown that the method is efficient and is more accurate than a number of alternative methods.
The method works best with torsion angles as coordinates. Note that this method calculates a relative rather than an "absolute" entropy; i.e. only entropy differences are physically meaningful.
We have demonstrated the power of the method by calculating the entropy differences between various disulfide-bonded forms of tachyplesin, a 17-residue peptide (see figure above); and we also successfully applied it to BPTI, a 58-residue protein.
The method works in full-dimensional configuration space which limits the size of the molecules to which it is applicable, but it can also be used as part of the mutual information expansion approximation, thus allowing its use for large molecules as well.
The success of the method is due to two key factors: (1) The use of the very efficient greedy learning algorithm by Verbeek et al. (2003), (2) The use of a cross-validation based stopping criterion, which efficiently prevents both underfitting and overfitting.
In addition to calculating an entropy, the method also provides an analytical model of the configurational probability density function, which can have a number of other uses besides entropy calculation.
We have implemented the method in Python/Numpy. The program is available for downloading at gmentropy.szialab.org.
Please see the following paper for more details:
|
Gergely Gyimesi, Péter Závodszky, and András Szilágyi: |